Innovating with Purpose to End AIDS | A Complete HIV-1 Whole-Genome Testing Solution
01 Why Has HIV-1 Whole-Genome Testing Become Central to AIDS Prevention and Control?
Human Immunodeficiency Virus (HIV) is the primary pathogen responsible for the global, severe infectious disease known as acquired immunodeficiency syndrome (AIDS). To this day, it remains a major challenge for global public health prevention and control. The core reason behind this challenge lies in HIV’s extremely high genetic variability and rapid evolutionary capacity. HIV uses RNA as its genetic material. During replication, it relies on reverse transcriptase, which lacks proofreading capability, resulting in a very high mutation rate. At the same time, the virus replicates rapidly within the host and undergoes frequent genetic recombination. Under the combined pressures of the immune system and drug selection, HIV quickly forms highly diverse quasispecies. This continuously evolving nature makes HIV like a cunning enemy that constantly “changes its form”: it can evade immune surveillance. It can rapidly develop drug resistance during treatment, leading to reduced efficacy or even treatment failure. The spread of drug-resistant strains, therefore, poses a long-term challenge to both clinical management and population-level prevention and control.
As HIV becomes increasingly evasive, how should we respond?
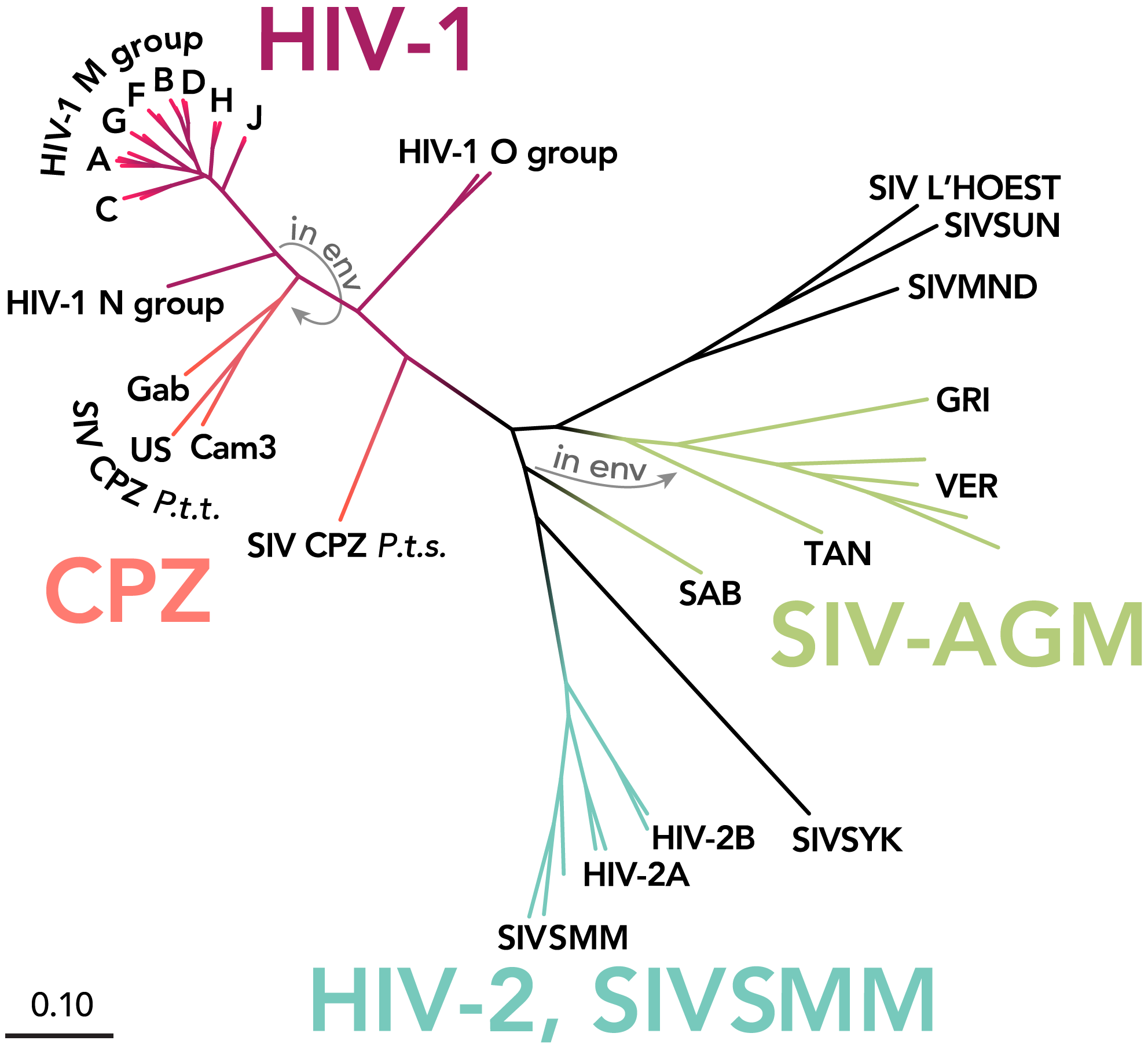
Based on genetic differences, HIV can be classified into two main subtypes, known as HIV-1 and HIV-2. HIV-1 is the predominant strain driving the global AIDS epidemic: it is more transmissible and more pathogenic, and its genetic diversity is the most complex. In contrast, HIV-2 is largely confined to West Africa and parts of Europe, with relatively lower transmission efficiency and pathogenicity. Within HIV-1, there are at least 10 genetic subtypes and circulating recombinant forms (CRFs). In China, the most widespread HIV-1 strains are CRF01_AE, CRF07_BC, and subtype B’. However, the composition of subtypes varies by region, and these distributions and epidemiological patterns directly influence the design of diagnostic approaches, treatment strategies, and prevention and control measures.
Against this backdrop, whole-genome testing for HIV-1 becomes particularly important. It provides an irreplaceable core value for AIDS prevention and control, enabling the precise identification of subtypes and recombinant forms, the rapid tracing of transmission chains, supporting clinical diagnostic decision-making, monitoring antiviral treatment responses, and accelerating the development of novel diagnostics, antiviral drugs, and vaccines.
Compared with conventional nucleic acid testing methods, the μCaler HIV-1 Whole-Genome Complete Testing Solution, powered by Nanodigmbio’s proprietary patented μCaler technology, offers significant advantages. The solution provides full coverage of the ~9.7 kb full-length HIV-1 genome, avoiding errors in assembly or scaffolding caused by repetitive sequences or recombination events. It is particularly strong in variant surveillance, precise genotyping, quasispecies identification, infection source tracing, and drug resistance analysis, providing robust technical support for the precision prevention and control of HIV infection cases as well as long-term standardized management.
02 HIV-1 Whole-Genome Testing Solution
2.1 Overview
μCaler HIV-1 Whole-Genome Complete Testing Solution is built on a EZ RNA & DNA Library Co-Preparation Kit and combines the μCaler Hybrid System with the μCaler HIV-1 Panel v1.0 to efficiently generate high-quality NGS libraries. It is compatible with mainstream sequencing platforms for sequencing and downstream analysis. Once sequencing is complete, the workflow—together with the XCapViz Bioinformatics Analysis Visual System and its online analysis portal—can deliver “sample in, results out” within a single day. This provides comprehensive technical support for accurate assembly of HIV-1 whole-genome sequences, subtype identification, and dynamic variant tracking, effectively enabling rapid, precise diagnosis and treatment for HIV infection cases as well as epidemiological surveillance and management.
In addition, leveraging Nanodigmbio’s flexible and versatile NadAuto series fully automated NGS workstations can further simplify laboratory workflows, reduce manual intervention, free up laboratory staff resources, increase clinical testing throughput and efficiency, and reduce human error and occupational exposure risk—providing an efficient and reliable solution to support epidemiological source-tracing studies and transmission risk assessment.
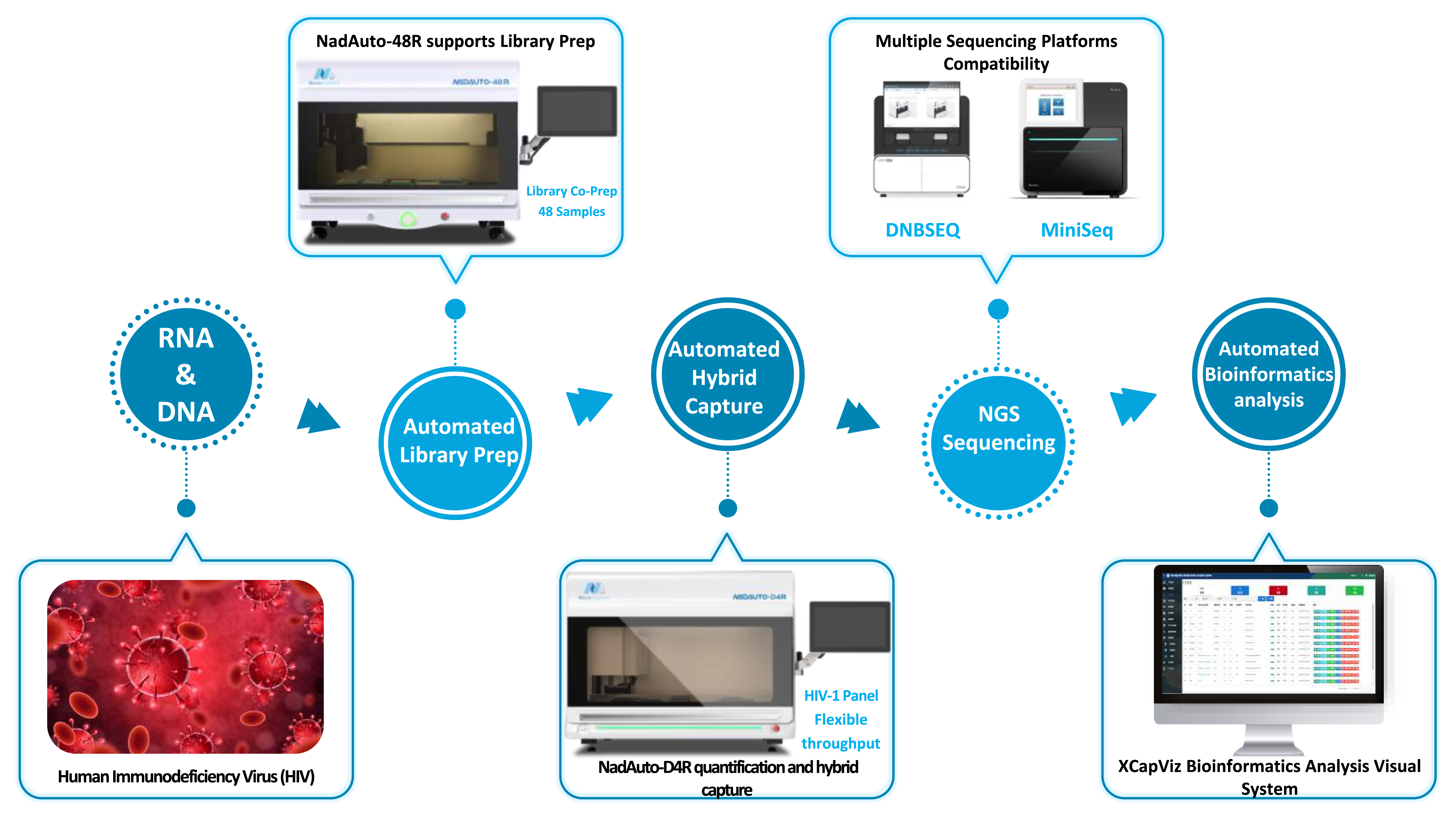
2.2 Key Highlights
• Ultra-fast workflow: 3-hour rapid library preparation (supports RNA-only sample input) and 1-hour ultra-fast hybridization, significantly improving NGS library preparation efficiency.
• Full-length coverage:Full-length genome probes designed based on reference sequences of globally prevalent HIV-1 strains, enabling complete resolution of the entire HIV-1 genome sequence.
• Precise analysis:Compared with single-/multi-gene genotyping/subtyping, whole-genome analysis more readily identifies complex circulating recombination events and provides more comprehensive annotation of drug-resistance sites.
03 Comprehensive HIV-1 Profiling, Precise Genotyping
3.1 Complete Whole-Genome Coverage
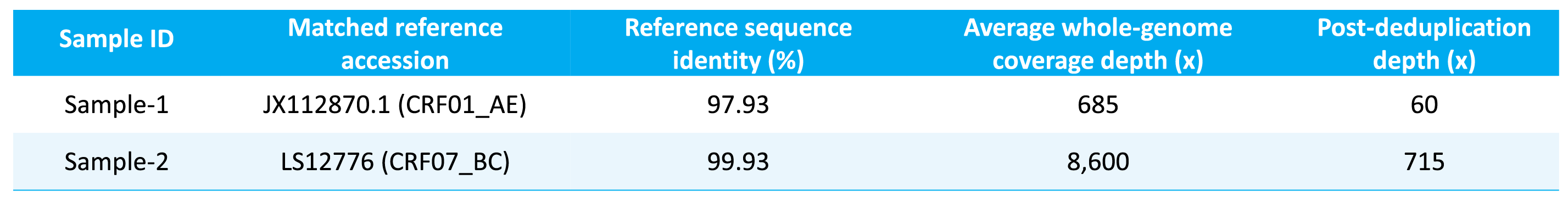
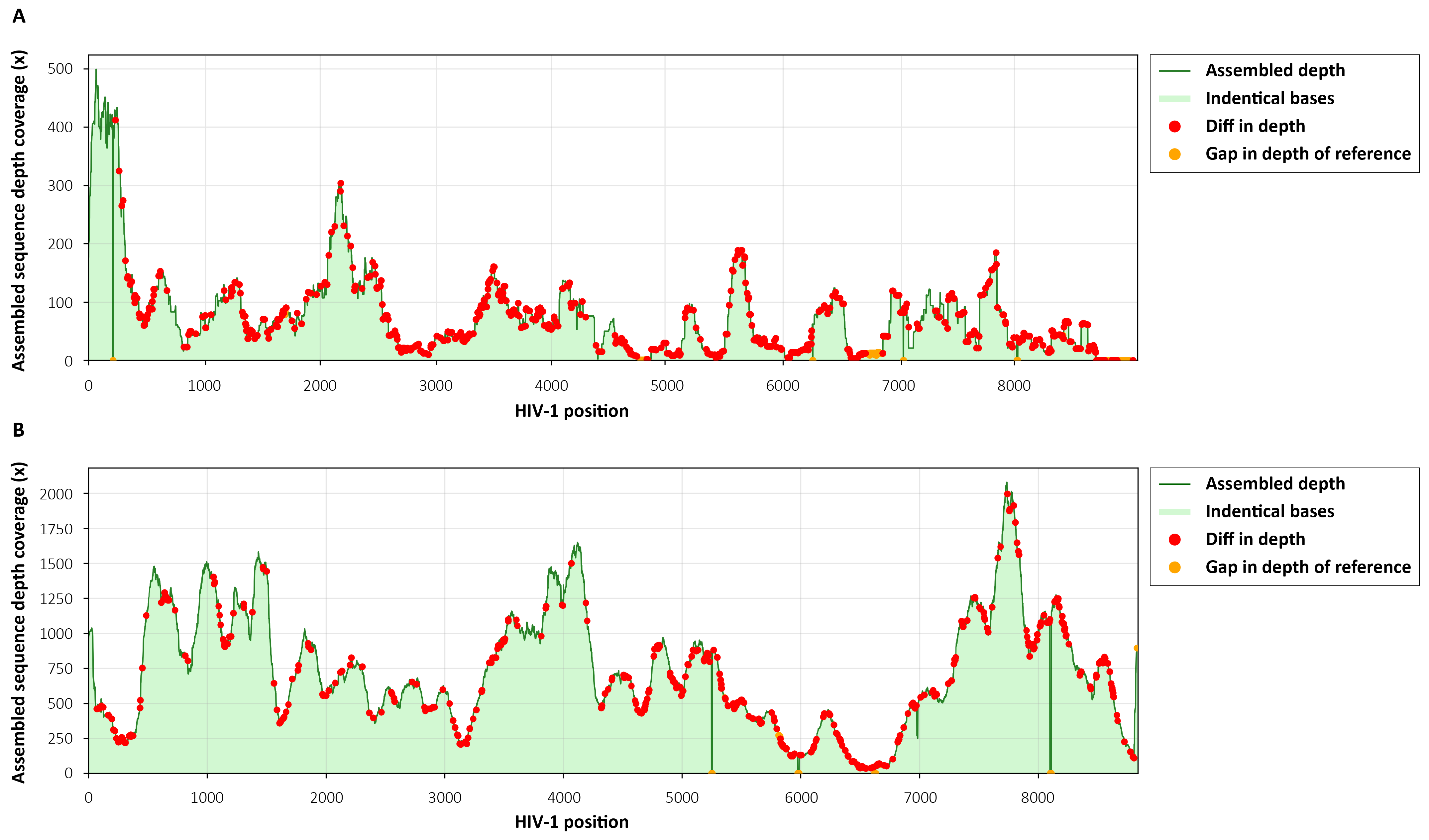
To evaluate the performance of the μCaler HIV-1 Whole-Genome Complete Testing Solution in HIV-1 whole genome sequencing and assembly, we analyzed the sequence identity between assembled full-length genome sequences and the reference sequence, and whole-genome coverage depth.
10 ng Carrier RNA (Cat # 1005601) was mixed with two clinically derived HIV nucleic acid samples. Pre-libraries were constructed using the EZ RNA & DNA Library Co-Preparation Kit. The capture was performed using μCaler Hybrid Capture Reagents v2 and μCaler HIV-1 Panel v1.0, followed by sequencing.
The results showed that the solution consistently generated high coverage of the full-length HIV-1 genome for different subtypes, with high concordance to the reference sequence (Table 1 and Figure 3). This provides a reliable data foundation for downstream in-depth variant analysis, CRF analysis, transmission chain tracing, and drug resistance annotation.
3.2 Precise Identification and Subtyping
HIV-1 can be classified into four subtype groups based on viral evolutionary characteristics: Group M (major), Group N (non-M, non-O), Group O (outlier), and the possibly existing Group P. Among them, Group M is widely prevalent worldwide, accounting for ~90% of global HIV infections. This group can be further subdivided into more than 9 subtypes (A, B, C, D, F, G, H, J, and K), as well as numerous CRFs and unique recombinant forms (URFs), forming an extremely complex spectrum of genetic diversity. From a genome-structure perspective, HIV-1 structural genes include pol, env, and gag, all of which can be used for subtype identification. Currently, the most comprehensive way to obtain subtype information is sequencing the full-length HIV-1 genome or partial gene regions (such as pol, env, and gag). Due to extensive drug resistance research and clinical monitoring, the pol region has become the most commonly used region for sequencing and subtyping analysis.
μCaler HIV-1 Whole-Genome Complete Testing Solution, combining full-length genome sequencing with pol gene analysis, has successfully identified multiple subtypes and CRFs, including: B, C, CRF01_AE, CRF07_BC, CRF08_BC, CRF55_01B, CRF59_01B, CRF65_cpx, CRF68_01B, and CRF85_BC. Subtype analysis was demonstrated using two clinically derived HIV nucleic acid samples (Table 2). The results showed that, for some samples, the subtype assignment based on the full-length genome sequence was inconsistent with the result based only on the pol gene. Such discrepancies typically suggest that the strain may be a multi-subtype recombinant virus.

To further validate putative sites of recombination, aligned segments were then analyzed by REGA HIV-1 Subtyping Tool (Version 3.0) in Sample-2. The recombination pattern plot showed that this strain’s genome is formed by an alternating pattern of sequence segments derived from different parental subtypes. Bootscan analysis further confirmed that, across different genomic regions, the bootstrap values (breakpoints support value) for the strain relative to reference sequences of potential parental subtypes exhibited a clear change. The sites of specific breakpoints were highly consistent with the recombination breakpoints predicted in the pattern plot. The high bootstrap confidence observed in multiple regions (typically > 70%) statistically substantiated the subtype assignment of each genomic segment (Figure 4).
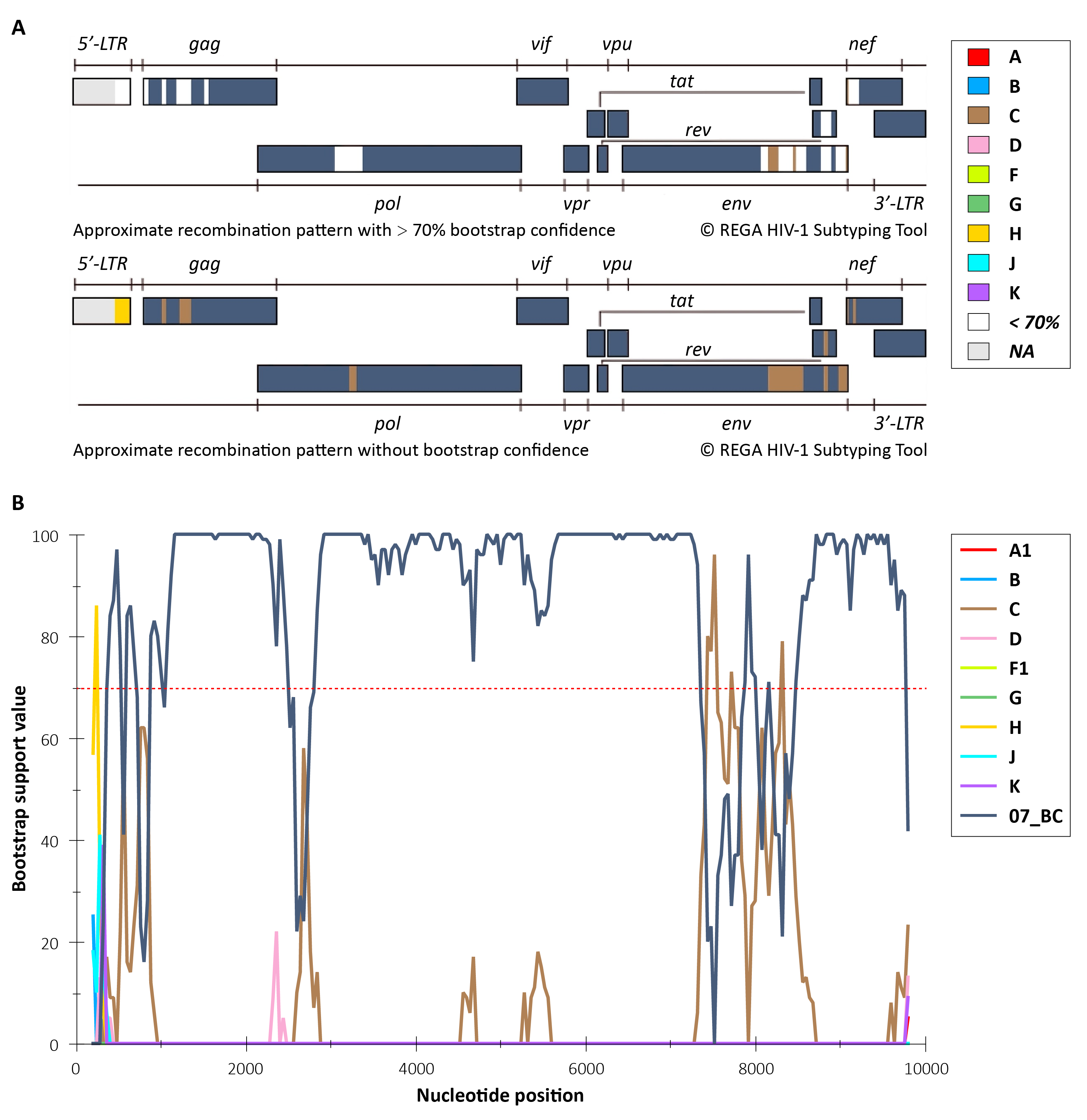
These results indicate that μCaler HIV-1 Whole-Genome Complete Testing Solution can more comprehensively and accurately reveal the virus’s complex recombinant background. In contrast, analyses that rely only on a single gene fragment region (such as pol) may fail to fully capture these recombination features. Therefore, rapid and accurate whole-genome subtyping and identification are of great importance for HIV-1 molecular epidemiology management, antiretroviral therapy monitoring, and global AIDS prevention and control.
04 Conclusion and Future Directions
μCaler HIV-1 Whole-Genome Complete Testing Solution, built on EZ RNA & DNA Library Co-Preparation Kit, a proprietary patented μCaler Hybrid System, and the XCapViz Bioinformatics Analysis Visual System with an online analysis portal, enables high-coverage, systematic profiling of complex and highly diverse full-length HIV-1 genomes. It accurately identifies multi-subtype recombination events, significantly improves the accuracy of subtype assignment and molecular source-tracing capability, and provides a reliable and efficient toolset to support transmission chain tracing, annotation of drug-resistance mutations, and clinical decision-making.
Looking ahead, as HIV molecular epidemiological surveillance continues to evolve toward more detailed and systematic approaches, and as demand for personalized treatment grows, the μCaler HIV-1 Whole-Genome Complete Testing Solution is expected to deliver greater value across diverse use cases—including the development of disease surveillance networks, drug-resistance risk assessment, and translation of research outputs. It will provide sustained, robust technical support for long-term precision prevention and control of AIDS, helping advance the global goal of ending the AIDS epidemic.
Solutions
- Methyl Library Preparation Total Solution
- Sequencing single library on different platform--Universal Stubby Adapter (UDI)
- HRD score Analysis
- Unique Dual Index for MGI platforms
- RNA-Cap Sequencing of Human Respiratory Viruses Including SARS-CoV-2
- Total Solution for RNA-Cap Sequencing
- Total Solution for MGI Platforms
- Whole Exome Sequencing
- Low-frequency Mutation Analysis
Events
-
Overseas Exhibition Preview | Nanodigmbio Looks Forward to Meeting You at ESHG Gothenburg, Sweden

-
Exhibition Preview | Nanodigmbio invites you to join us at Boston 2025 Annual Meeting of the American Society of Human Genetics (ASHG)

-
Exhibition Preview | Nanodigmbio Invites You to Join Us at WHX & WHX Labs Kuala Lumpur 2025, Malaysia International Trade and Exhibition Centre in Kuala Lumpur

-
Exhibition Preview | Nanodigmbio Invites You to Join Us at Hospitalar 2025, Brazil International Medical Device Exhibition in São Paulo

-
Exhibition Preview | Nanodigmbio invites you to join us at Denver 2024 Annual Meeting of the American Society of Human Genetics (ASHG)

-
Exhibition Preview | Nanodigmbio invites you to join us at Sapporo 2024 Annual Meeting of the Japan Society of Human Genetics (JSHG)
