冠状病毒变异到什么地步了?μCaler 精准追踪!
新知小课堂
Q 哪些病原体会导致普通感冒?
最常见的是鼻病毒,其次是冠状病毒、副流感病毒、呼吸道合胞病毒、腺病毒、柯萨奇病毒、埃可病毒等。此外,人偏肺病毒、肺炎支原体和肺炎衣原体也可能引起感冒样症状。这些病原体通过飞沫或接触传播,导致鼻塞、流涕、咽痛等症状,大多为自限性。但如出现呼吸困难、持续高热等情况,应及时就医。
01 背景
人类冠状病毒 (HCoVs) 是呼吸道感染的重要病原。其中,SARS-CoV、MERS-CoV 和 SARS-CoV-2 属高致病性病毒,可致重症甚至死亡;而 HCoV-229E、HCoV-OC43、HCoV-NL63、HCoV-HKU1 多表现为普通感冒样症状,但在婴幼儿、老年人和免疫力低下者中也可导致严重肺炎。
冠状病毒拥有 RNA 病毒中最大的基因组 (约 30 kb),且极易发生突变和重组,特别是刺突蛋白 (Spike) 上的变化会影响免疫逃逸和跨物种传播。因此,持续监测其演化对公共卫生至关重要。
然而,传统测序方法存在操作复杂、周期长、对低载量样本灵敏度低等问题。为突破瓶颈,江苏省疾控中心基于纳昂达独家专利的 μCaler® 技术,研发了新型微靶捕获测序技术 (MT-Capture),并对 SARS-CoV-2 等冠状病毒进行了全基因组测序,相关成果于 2024 年 4 月发表在 mSystem (IF=6.4) 期刊上。
2025 年 4 月,江苏省疾控中心等单位在 Archives of Virology 期刊上发表了题为“Genetic characterization of coronaviruses causing common cold symptoms based on micro-targeted capture sequencing”的研究性论文。该研究聚焦四种“感冒型”人类冠状病毒,利用 MT-Capture 完成对 14 例临床样本的全基因组测序,深入解析其演化特征。
该研究中文库构建与杂交捕获方案由纳昂达提供,
纳昂达为高质量病毒全基因组测序研究提供了关键基础。
02 研究方法
2.1 样本来源
2017-2023 年,江苏省疾控中心及省内医院收治的 14 例呼吸道感染患者咽拭子,经 qPCR 鉴定分型:HCoV-229E (2 例)、HCoV-HKU1 (3 例)、HCoV-NL63 (4 例)、HCoV-OC43 (5 例),Ct 值 23-36。
2.2 病毒全基因组测序
基于 μCaler® 技术,MT-Capture 针对四类病毒的参考基因组设计特异性探针,并搭配 μCaler® 杂交捕获系统中的 μCaler® NanoBlockers (#1106101) 和 μCaler® Hybrid Capture Reagents v2 (#1105201) 完成捕获文库构建,最终在 MiniSeq 平台完成 PE150 测序。
2.3 数据分析
下机数据经 Fastp 0.21.0 质控后,利用 IRMA v1.0.3 完成 HCoV 全基因组拼接与覆盖度评估;再基于参考基因组通过 BWA 0.7.17 比对并用 bcftools 1.11 识别 SNP,结合 snpEff 5.0e 注释功能影响;随后采用 MAFFT 与 MEGA 11 构建刺突蛋白基因系统进化树,确定样本基因分型;最后使用 NGlyc 1.0 预测刺突蛋白 N-糖基化位点,揭示关键突变与糖基化模式变异。
03 研究结果
3.1 MT-Capture 技术灵敏度
该研究通过参考品梯度稀释实验系统评估了 MT-Capture 技术的灵敏度。结果显示,该技术在 Ct ≤ 32 的样本中可实现 100% 全基因组覆盖,在更高 Ct 值 (低病毒载量) 样本中仍表现优异:Ct=35 的 HCoV-HKU1 覆盖率达 94.73%,Ct=37 的 HCoV-NL63 覆盖率仍达 85.25%,且所有样本测序深度均匀 (图 1.),充分验证该方法对低病毒载量临床样本的检测能力远超传统方法。
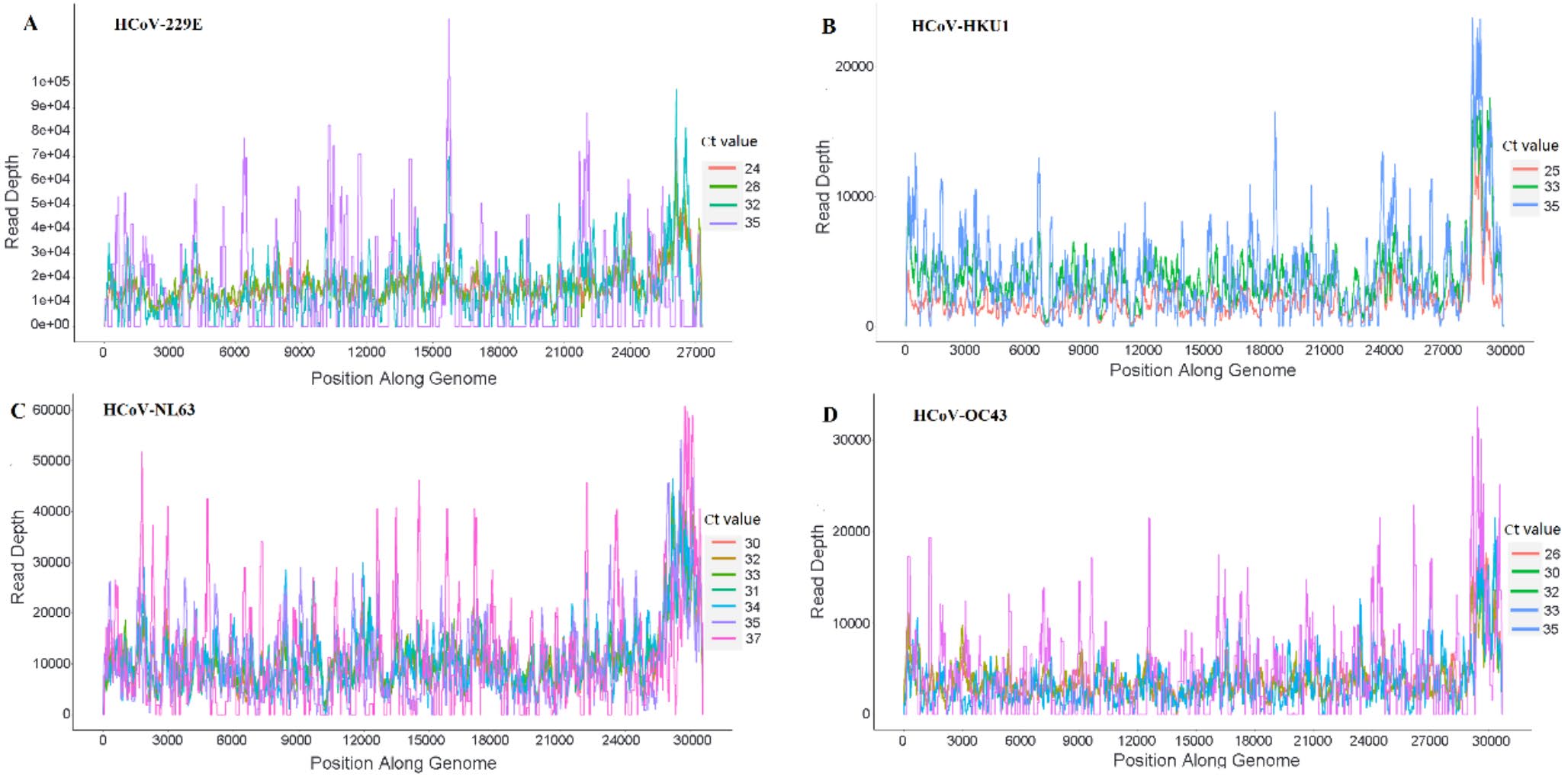
图 1. MT-Capture 对不同 Ct 值 HCoV 病毒基因组的覆盖范围和测序深度。
3.2 病毒进化分析
通过刺突蛋白基因系统进化树分析,该研究系统绘制了四种人类冠状病毒的演化格局。结果显示,江苏地区四种临床毒株均与全球流行基因型保持一致:HCoV-229E 属基因群 6,HCoV-HKU1 以 A 型为主并伴随 C 型;HCoV-NL63 涉及 B、C2、C3 型,而 HCoV-OC43 呈现 I 型与新兴 K 型共存 (图 2. A-D)。同时,全基因组扫描发现多处关键功能位点突变 (如 Y406G、K478N、I507L、N483D),这些变异可能影响受体结合、免疫逃逸及病毒适应性,提示持续的分子监测对公共卫生防控与疫苗药物研发至关重要。
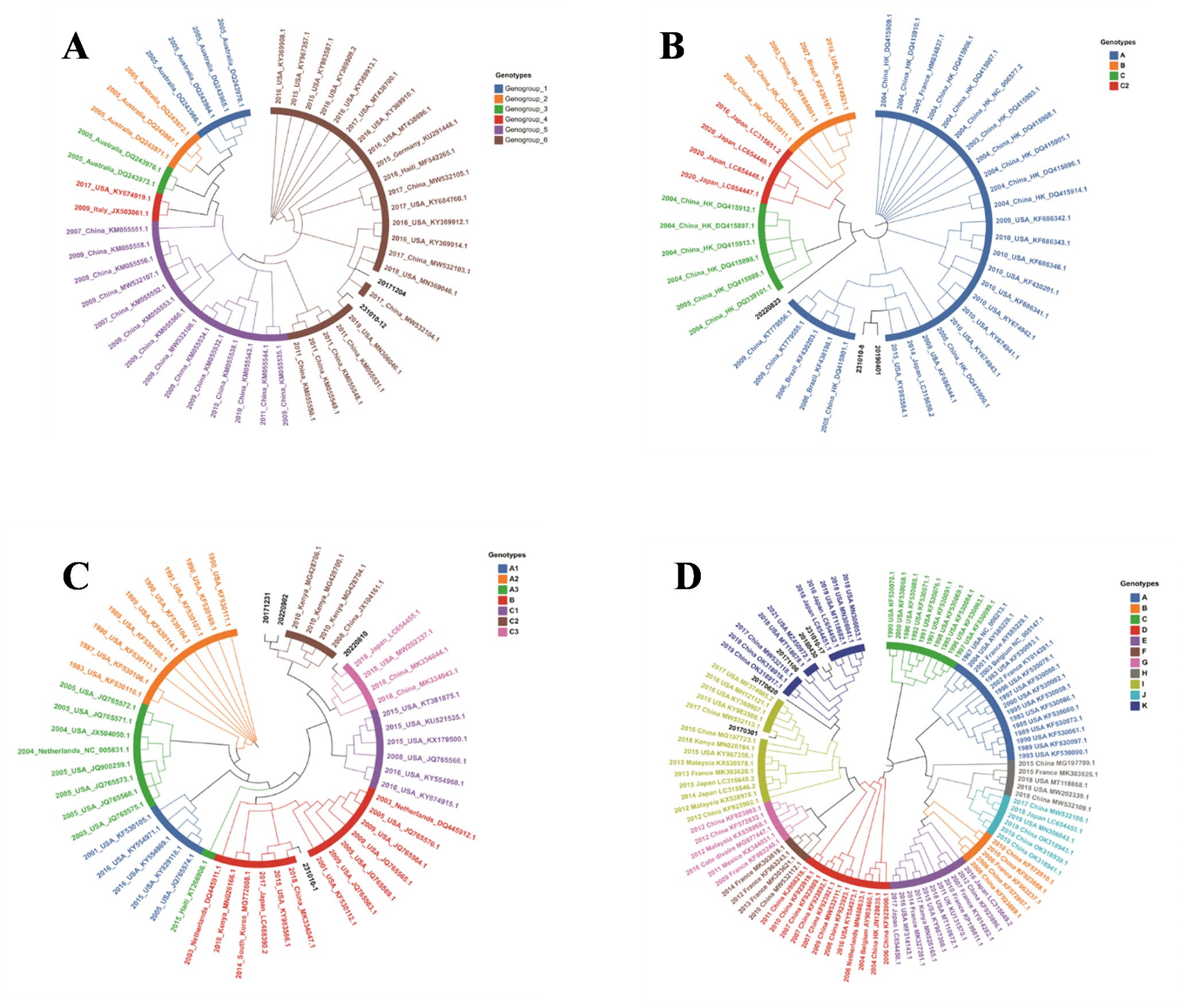
图 2. 基于四种 HCoV 的 S 基因构建的进化树。
3.3 刺突蛋白的氨基酸变异与糖基化位点预测
在刺突蛋白编码基因的分析中,该研究鉴定出多处受体结合域 (RBD) 的关键突变:如 HCoV-229E 的 Y406G 可能导致受体结合环电荷重新分布,HCoV-NL63 的 S267F 及 HCoV-OC43 的 T246I/N483D 或将直接改变 RBD 构象并影响其与 ACE2 的结合能力,而 HCoV-OC43 的 H124Y 则可能重塑神经氨酸酯酶活性域功能。
进一步的糖基化预测揭示了病毒免疫逃逸的重要机制:HCoV-OC43 K 型在 RBD 侧翼新增 S81N、T113N 等 4 个 N-糖基化位点,或形成空间屏障以遮蔽中和表位;相反,HCoV-HKU1 的 N74 位点丢失则降低了糖基化密度。前者体现了“糖基化屏障扩张”,后者则体现了“精简演化”,两种策略均有助于病毒适应宿主免疫环境。
这些结构域特异突变与糖基化重编程相互作用,共同优化了病毒的宿主适应性,并为未来干预靶点设计提供了分子依据。
04 总结与展望
该研究基于纳昂达 μCaler® 技术建立的 MT-Capture 技术,突破了低病毒载量 HCoV 临床样本全基因组解析的瓶颈,系统绘制了江苏地区四种人类冠状病毒的分子流行病学图谱。同时,研究揭示了刺突蛋白受体结合域的关键突变与糖基化位点的动态重编程,为冠状病毒跨物种传播机制提供了新的理论依据。所鉴定的功能性突变及修饰位点可成为新型疫苗与广谱抗病毒药物的优先靶标。该技术也展示出拓展至更多类型病原体检测领域的潜力,高灵敏度全基因组测序结合进化追踪与功能验证研究,将为新发突发传染病的精准预警和干预策略提供有力支撑。
参考文献
[1] Qiao Q, Zhu X, Meng Z, et al. Genetic characterization of coronaviruses causing common cold symptoms based on micro-targeted capture sequencing[J]. Archives of Virology, 2025, 170(5): 98.





